4.Análisis de datos NGS
…
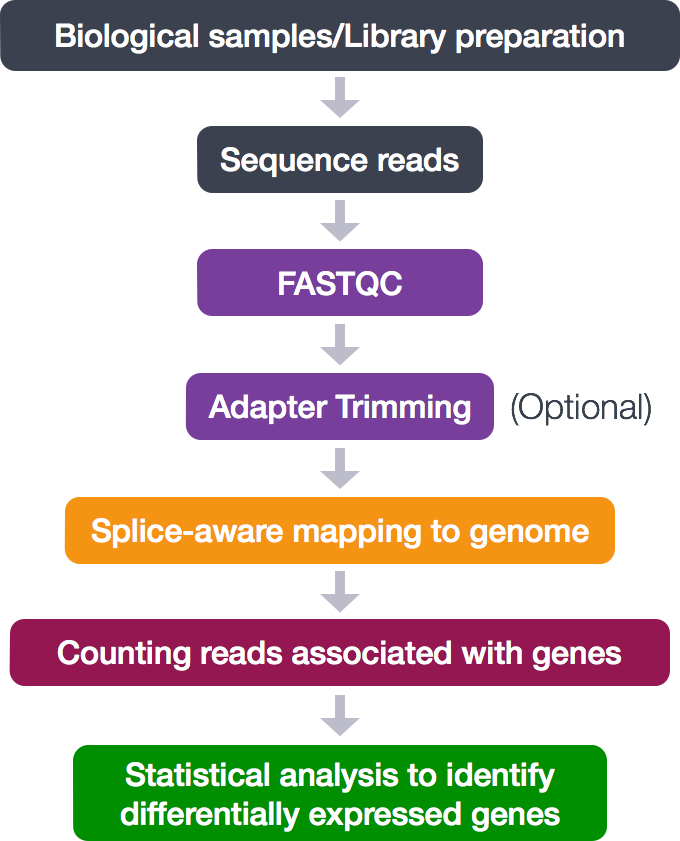
CONTENIDO
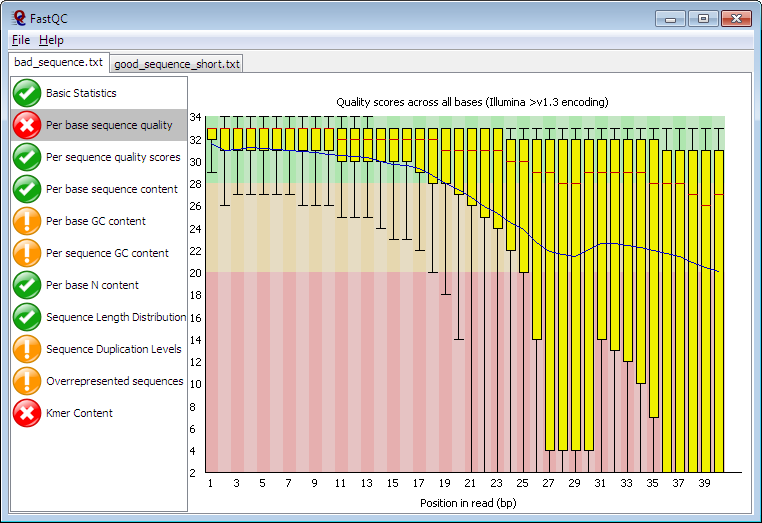
CONTROL CALIDAD
fastqc -t 2 cavtsc_forward_paired.fq.gz cavtsc_reverse_paired.fq.gz -o /mnt/disco2/fascue/cporcellus/results/fastqc/
FILTRADO DE READS
java -jar ../descargas/Trimmomatic-0.39/trimmomatic-0.39.jar PE
-phred33
-threads 2
file_1.fastq file_2.fastq
file_forward_paired.fq.gz file_forward_unpaired.fq.gz
file_reverse_paired.fq.gz file_revers_unpaired.fq.gz
ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10
LEADING:3
TRAILING:3
SLIDINGWINDOW:4:25
MINLEN:25
java -jar ../descargas/Trimmomatic-0.39/trimmomatic-0.39.jar PE -phred33 -threads 2 file_1.fastq file_2.fastq file_forward_paired.fq.gz file_forward_unpaired.fq.gz file_reverse_paired.fq.gz file_revers_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:25 MINLEN:25
java -jar ../descargas/Trimmomatic-0.39/trimmomatic-0.39.jar SE
-phred33
-threads 2
file.fastq
file_trimm.fq
ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:10
LEADING:3
TRAILING:3
SLIDINGWINDOW:4:25
MINLEN:25
ILLUMINACLIP:<fastaWithAdaptersEtc>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>
LEADING:<quality>
TRAILING:<quality>
SLIDINGWINDOW:<windowSize>:<requiredQuality>
MINLEN:<length>
ALINEAMIENTO

#cargar paquetes para R
library(Rbowtie2)
library(Rsamtools)
library(ape)
library(viridisLite)
library(viridis)
Descargar secuencia de referencia Genoma de A. thaliana , para tener un panorama del organismo en el que estamos trabajando se debe analizar el genoma anotado, para ello podemos trabajar en R con archivos gff
# read gff files with ape
gff_file <- read.gff("sequence.gff3", na.strings = c(".", "?"), GFF3 = TRUE)
# transform in matrix to filter annotations
tab <- as.matrix(table(gff_file$type))
rnames <- as.matrix(rownames(tab))
# make a plot of annotation feactures
etiquetas <- paste0(rnames[c(4,7,10,16),],"=",round(100 * tab[c(4,7,10,16),]/sum(tab[c(4,7,10,16),]), 2), "%")
par(mfrow=c(1,2), adj = TRUE)
pie(tab[c(4,7,10,16),], labels = etiquetas, col = viridis(4))
#pie(tab[c(4,7,10,16),], col = viridis(4), labels = paste0(tab[c(4,7,10,16),], "%"))
barplot(tab[c(4,7,10,16),], col = viridis(4), width = 60)
Preparacion del index
bowtie2_build("AthalianaChr4.fasta", bt2Index = "index/" , overwrite = TRUE)
Alineamiento de secuencias
bowtie2_build("AthalianaChr4.fasta", bt2Index = "index/" , overwrite = TRUE)
bowtie2(bt2Index = "index/",
samOutput = "SRR390310.sam",
seq1 = "SRR390310_1.fastq",
seq2 = "SRR390310_2.fastq",
"--threads=3")
Convertir SAM a BAM
asBam("SRR390310.sam")
Visualizar alineamiento
Indexar el genoma con STAR
| Parámetro | Descripción |
|---|---|
--runMode | indica el tipo de opcion que utilizará STAR, en este caso queremos generar un índice del genoma por lo que utilizamos la flag genomeGenerate |
--genomeDir | indica donde se guardaran los resultados del indice y la ubicación de los archivos del genoma |
--genomeFastaFiles | indica donde estan almacenadas las secuencias del genoma en formato FASTA |
--sjdbGTFfile | sj: splice junction db: database GTFfile: archivo GTF indica la ubicación del archivo GTF para mejorar e improvisar el mapeo dado el modelo de los genes |
--sjdbOverhang | Especifica el largo a considerar de la secuencia genómica alrededor del splice junction, este valor esta ligado al largo de los reads y deberia ser max(ReadLength) - 1 |
--runThreadN | total de hebras que se ejecutaran en paralelo, este número no debe sobrepasar la cantidad de cores que tiene un computador y pruebas de escalamiento deberian ser ejecutadas para calcular el óptimo |
STAR \
--runMode genomeGenerate \
--genomeDir genome/star_index \
--genomeFastaFiles genome/NC_000021.9.fna \
--sjdbGTFfile annotation/NC_000021.9.gtf \
--runThreadN 2
STAR --runMode genomeGenerate --genomeDir genome/star_index/ --genomeSAindexNbases 7 --genomeFastaFiles genome/NC_000932.1.fasta --sjdbGTFfile genome/NC_000932.1.gtf --runThreadN 2
Alinear el genoma
| Parámetro | Descripción |
|---|---|
--readFilesIn | archivo de reads a mapear |
--genomeDir | indica donde esta alojado el genoma |
--runThreadN | cantidad de threads |
--outSAMType | tipo de archivo SAM o BAM en este caso indicamos BAM |
SortedByCoordinate | Ordenar el archivo BAM por las coordenadas del genoma |
--outFileNamePrefix | Prefijo para los archivos de salida |
Command
# Help
STAR -h
# Run STAR (~3min)
STAR \
--genomeDir genome/star_index \
--readFilesIn results/trimmed/sample_filtered.fq \
--runThreadN 2 \
--outSAMtype BAM SortedByCoordinate \
--quantMode GeneCounts
STAR --genomeDir ../../../genome/star_index/ --readFilesIn ../../trimmed/NC_000932.1_6_trimmed.fq --runThreadN 2 --outSAMtype BAM SortedByCoordinate --limitBAMsortRAM 12000000000 --quantMode GeneCounts